Epidermolytic ichthyosis
| Epidermolytic Ichthyosis (EI) | |
|---|---|
| Other names: Bullous epidermis ichthyosis, bullous epidermis ichthyosis (BEI), bullous congenital ichthyosiform erythroderma (BCIE),[1] bullous ichthyosiform erythroderma[2] bullous congenital ichthyosiform erythroderma Brocq[3] | |
.jpg) | |
| Redness, blisters | |
| Specialty | Dermatology |
| Symptoms | Early: Red skin, blisters, skin breakdown, peeling[4] Later: Thickened skin, itch, fissures, absence of sweat, strong body odor[5] |
| Complications | Infection, dehydration, electrolyte imbalance, joint problems[5] |
| Usual onset | Newborn[4] |
| Duration | Lifelong[5] |
| Types | With and without involvement of palms and soles[5] |
| Causes | Mutations in keratin 1 or keratin 10[5] |
| Risk factors | Family history[5] |
| Diagnostic method | Appearance, skin biopsy, genetic testing[5] |
| Differential diagnosis | Other causes of ichthyosis[5] |
| Prevention | Pre-natal screening[5] |
| Treatment | Moisturisers, retinoid[4] |
| Medication |
|
| Prognosis | Less severe with age[4] |
| Frequency | Rare, males = females[5] |
| Deaths | Unknown[5] |
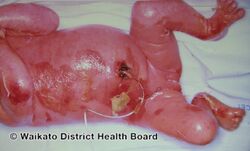
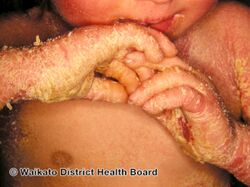
Epidermolytic ichthyosis (EI), formerly known as epidermolytic hyperkeratosis, is a severe form of dry scaly skin, that starts with redness, blisters, skin breakdown, and peeling in the newborn.[4][5] Excessive cornification (thickening) of skin typically develops several months later.[5] Other symptoms include itch, painful fissures, strong body odor, and absence of sweat.[5] Severity and extent of skin involvement vary.[4] Complications may include dehydration, infection including sepsis, joint problems, and electrolyte imbalance.[5]
It is caused by a genetic mutation in the gene encoding the proteins keratin 1 or keratin 10, resulting in disruption of the structure of the outermost layer of the skin.[5] The condition is mostly inherited in an autosomal dominant pattern.[5] To a lesser extent, a recessive form exists.[4] Diagnosis is by its appearance, skin biopsy, and genetic testing.[5] The two main types are divided into one involving palms and soles and the other without.[5]
Treatment includes regularly applying thick moisturisers.[4] Other therapies include retinoids, applied to the skin as creams or taken by mouth, N-acetylcysteine, liarozole, and calcipotriol.[4][5] Antibacterial soaps, chlorhexidine, and dilute sodium hypochlorite baths may reduce secondary bacterial colonization of skin.[5] The severity of the condition generally lessens with age.[4]
Epidermolytic ichthyosis is rare, affecting around 1 in 200,000 to 300,000 newborns.[5] Males and females are affected with equally frequency.[5] The condition was first described in 1902 by Brocq.[6] It was first classified by its presence or absence in the palms and soles by DiGiovanna and Bale in 1994.[5][7] Those affected often socially isolated due to the condition.[8]
Signs and symptoms
EI initially typically presents with redness, blisters, erosions, and peeling in a newborn baby.[5] Because newborns with this disorder are missing the protection provided by normal skin, they are at risk of becoming dehydrated and developing infections in the skin or throughout the body (sepsis).[5]
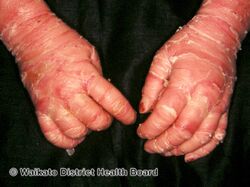
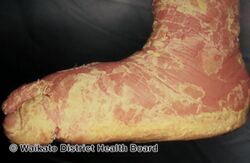
As affected individuals get older, blistering is less frequent, red skin becomes less evident, and the skin becomes thick (hyperkeratotic), especially over joints, on areas of skin that come into contact with each other, or on the scalp or neck. This thickened skin is usually darker than normal. Bacteria can grow in the thick skin, often causing a distinct odor.[5]
Complications include infection and joint problems.[5] Affected newborns are particularly at risk of dehydration, sepsis, and electrolyte imbalance.[5]
- Baby
-
Epidermolytic hyperkeratosis
-
Epidermolytic hyperkeratosis
-
Epidermolytic hyperkeratosis
-
Epidermolytic hyperkeratosis
-
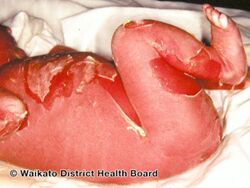
Epidermolytic hyperkeratosis
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
- Adult
-
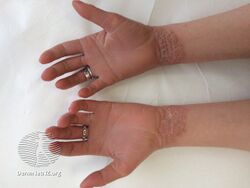
Epidermolytic hyperkeratosis
.jpg)
Cause and mechanism
This is a dominant[9] genetic condition caused by mutations in the genes encoding the proteins keratin 1 or keratin 10.
- Keratin 1 is associated with the variants affecting the palms and soles.
- Keratin 10 is associated with the variants in which these are unaffected.
Diagnosis
The condition can be diagnosed via exam that reveals; generalized redness; thick, generally dark, scales that tend to form parallel rows of spines or ridges, especially near large joints; the skin is fragile and blisters easily following trauma; extent of blistering and amount of scale is variable.
Classification
EI is a severe form of dry scaly skin and can be categorized into two main types :-
- People with PS-type epidermolytic hyperkeratosis have thick skin on the palms of their hands and soles of their feet (palmoplantar or palm/sole hyperkeratosis) in addition to other areas of the body.[5]
- People with the other type, NPS-type, do not have extensive palmoplantar hyperkeratosis, but do have hyperkeratosis on other areas of the body.[5]
Treatment
Treatment includes applying thick moisturisers.[4] Other therapies include topical and oral retinoids.[4] These include topical N-acetylcysteine, liarozole, and calcipotriol.[5] Antibacterial soaps, chlorhexidine, and dilute sodium hypochlorite baths may reduce secondary bacterial colonisation of skin.[5]
Epidemiology
The condition is rare, affecting around 1 in 200,000 to 300,000 babies.[5]
History
EI was first classified by its presence or absence in the palms and soles by DiGiovanna and Bale in 1994.[5][7]
Research
Gene therapy is being studied for EI.[10]
See also
- Ichthyosis bullosa of Siemens
- List of cutaneous conditions caused by mutations in keratins
- Nonbullous ichthyosiform erythroderma
References
- ↑ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- ↑ Fitzpatrick's dermatology in general medicine (6th ed.). New York: McGraw-Hill, Medical Pub. Division. 2003. p. 482. ISBN 0-07-138076-0.
- ↑ "Brocq's disease II". www.whonamedit.com. Archived from the original on 9 December 2021. Retrieved 21 May 2024.
- ↑ 4.00 4.01 4.02 4.03 4.04 4.05 4.06 4.07 4.08 4.09 4.10 4.11 James, William D.; Elston, Dirk; Treat, James R.; Rosenbach, Misha A.; Neuhaus, Isaac (2020). "27. Genodermatoses and congenital anomalies". Andrews' Diseases of the Skin: Clinical Dermatology (13th ed.). Edinburgh: Elsevier. pp. 563–565. ISBN 978-0-323-54753-6. Archived from the original on 2023-08-05. Retrieved 2024-05-19.
- ↑ 5.00 5.01 5.02 5.03 5.04 5.05 5.06 5.07 5.08 5.09 5.10 5.11 5.12 5.13 5.14 5.15 5.16 5.17 5.18 5.19 5.20 5.21 5.22 5.23 5.24 5.25 5.26 5.27 5.28 5.29 5.30 5.31 5.32 5.33 5.34 5.35 5.36 Rice, Ashley S.; Crane, Jonathan S. (2023). "Epidermolytic Hyperkeratosis". StatPearls. StatPearls Publishing. PMID 31335043. Archived from the original on 2024-05-19. Retrieved 2024-05-19.
- ↑ Kwak, Juliann; Maverakis, Emanual (2006). "Epidermolytic hyperkeratosis". Dermatology Online Journal. 12 (5). doi:10.5070/D38HV5R331. ISSN 1087-2108. Archived from the original on 31 January 2023. Retrieved 21 May 2024.
- ↑ 7.0 7.1 DiGiovanna JJ, Bale SJ (August 1994). "Clinical heterogeneity in epidermolytic hyperkeratosis". Arch Dermatol. 130 (8): 1026–35. doi:10.1001/archderm.130.8.1026. PMID 8053700.
- ↑ "Epidermolytic ichthyosis | DermNet". dermnetnz.org. Archived from the original on 9 June 2023. Retrieved 21 May 2024.
- ↑ Ross R, DiGiovanna JJ, Capaldi L, Argenyi Z, Fleckman P, Robinson-Bostom L (July 2008). "Histopathologic characterization of epidermolytic hyperkeratosis: a systematic review of histology from the National Registry for Ichthyosis and Related Skin Disorders". J. Am. Acad. Dermatol. 59 (1): 86–90. doi:10.1016/j.jaad.2008.02.031. PMC 2517215. PMID 18571597.
- ↑ Joosten, M. D. W.; Clabbers, J. M. K.; Jonca, N.; Mazereeuw-Hautier, J.; Gostyński, A. H. (15 July 2022). "New developments in the molecular treatment of ichthyosis: review of the literature". Orphanet Journal of Rare Diseases. 17 (1): 269. doi:10.1186/s13023-022-02430-6. ISSN 1750-1172. PMC 9287901. PMID 35840979.
External links
| Classification | |
|---|---|
| External resources |